PARASOL
Thin film growth and surface erosion: Both processes are based on particles impacting on a surface. In surface erosion typical particle energies are keV. In thin film formation the energy is mostly thermal, even in cases of ion assisted or sputter deposition also energetic particles will hit the surface. Thus both processes can be studied by the Molecular Dynamics (MD) technique.
In MD the evolution of a system of N mutually interacting particles is calculated by numerical integration of the classical equations of motion. Thus MD allows in principle to describe the true dynamics of a system at a given temperature assuming the validity of classical mechanics. Essentially the only physics input is the proper choice of the interaction potential. In an MD calculation a particle is deposited at the surface and after a certain time another particle is started above the surface at a randomly chosen lateral position. This leads to the growth of a thin film or erosion, at low or high particle energies, respectively. The problem in a MD simulation of thin film deposition/erosion is the fact that for a calculation, which can be performed in a reasonable time of days or weeks even for the deposition/erosion of a few monolayers on a small crystal surface of the order of a few hundred atoms deposition/erosion rates will be in the order of meters per second. These are rates, which are by factor of at least 107 higher, than what can be achieved in any experiment. The main problem is the fact, that at RT during such a fast deposition/erosion all diffusion processes at the surface, with typical activation energies in the order of a few tenth of an eV will be suppressed, as they occur rather on a time scale of μs and not ps.
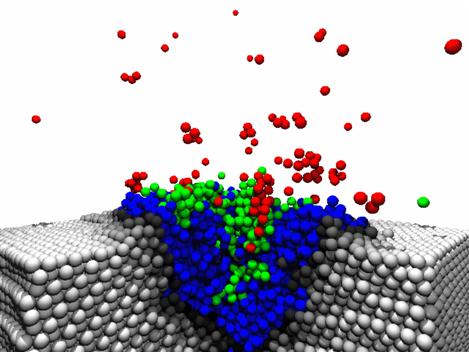
The picture shows the result of a Molecular Dynamics simulation of
5 keV Argon atom impact on a "soft" Cu(111) crystal. In the
simulation a "soft" Cu crystal with an artificially lowered surface
binding energy by 50% was used, which gives rise to a high
sputtering yield. The simulation results in emission of large
clusters, which become detached from the target after typically 5
ps. The picture shows the development of the impact crater after
2ps.
The color coding indicates temperature of the atoms: white - black
300K - 1400K; blue 1400K - 2800K; green 2800K - 4200K; red above
4200K.
Another numerical method which has been used to study thin film growth is the Kinetic Monte Carlo technique (KMC), which is a special variant of ordinary Monte Carlo (MC). KMC is a procedure for solving kinetic equations. Its aim is to reproduce faithfully a non-equilibrium or a relaxation process. The emphasis in this method is on a correct possible evolution of the system with time. Contrary to ordinary MC real time is included in the evolution of the system. With this technique diffusion of ad-atoms on a surface can be well described. For example an isolated ad-atom will jump to any of the four next local energy minima on a (100) surface with a rate, which is given by k(Eb,T) = k0 exp(-Eb/kbT), with Eb the potential barrier height (for a metal typically a few tenth of an eV). ko can be interpreted as an attempt frequency, i.e. the frequency an atom is oscillating around its equilibrium position at a given temperature and is in the order of 1012 – 1013s-1. In a similar manner on a crystal with a free surface we can describe all possible diffusion events. Thus we can follow the evolution of the system for a given time and then deposit a new particle at the surface and again continue with KMC for a given time.
After an energetic particle impact KMC is only possible after all collisional processes are over and the impact region has reached the target temperature. Thus KMC cannot be directly applied to surface erosion. But also deposition is not always occurring at thermal energies (vapor deposition) but quite often like in sputter deposition or in beam assisted deposition also more energetic atoms with energies of a few ten eV are deposited. The reason is, that the addition of energetic atoms to the deposited flux can have a considerable influence on the properties of the growing film, as for example island versus layer by layer growth. But even in the case of a purely thermal deposition beam, an atom will arrive at the surface with a kinetic energy of a few eV. This energy gain is due to the decrease in potential energy, if an atom is added to a crystal. The same amount of energy is necessary to remove an atom from the crystal surface and bring it infinitely far away (cohesive energy). Thus during the time in the order of a ps the region around the impact point will become quite hot and cannot be treated properly by KMC.
Thus to extend the time scale in MD calculations of erosion and deposition we have coupled an MD code to a KMC code. This combined MD-KMC code can be described in the following way:
- A MD calculation is performed for a crystal at a given temperature during the deposition of a single atom for a time tMD. tMD will depend on the energy of the deposited atom. In the case of thermal deposition a calculation time of 1 ps or even below is sufficient. In erosion tMD will be a few ps at low ion energies.
- At the end of the MD calculation all atoms receive their next nearest ideal lattice positions. If tMD is too small and the lattice has not fully relaxed, a lattice position might be occupied by either 2 or 0 atoms. An algorithm was developed to take care of this problem by extending or shrinking the lattice at this position. The drawback of this procedure is, that we also artificially remove all existing interstitials and vacancies formed in the bulk.
- Now a KMC calculation is started for a time tdiff, which can be up to seconds. Compared to an MD calculation for a few ps the used cpu time in a KMC calculation for seconds will still be short, at typical barrier heights of a few tenth of an eV and temperatures not too much above RT. The reason is that a diffusion step will occur on the μs – ms time scale and not on the fs time scale as in MD.
- The lattice at the end of the KMC step is heated up in an MD calculation to the target temperature and the next atom is deposited (back to step 1).
In the KMC calculations the different barrier heights for surface diffusion have been calculated by a quenched MD simulation using the same potentials as in the MD calculations.
Thus in the case of surface erosion as well as in sputter deposition the coupling of the MD calculation to a KMC calculation allows us to extend our calculations from a few ps to times of up to seconds until the next particle will impinge/be deposited on the crystal surface of about 100 nm2 in size. The latter value of 1 s is quite realistic for a typical sputter erosion or deposition experiment. In such a calculation thermal diffusion processes at the surface and annealing of the surface after energetic ion bombardment can be taken into account.
Results sofar have been obtained for surface erosion of Cu(100) under 200 – 600 eV Cu ion bombardment and for the growth of Cu on Cu(100) for deposition at thermal energies like in film formation by evaporation up to energies of 100 eV per atom like in sputter deposition or beam assisted deposition.
Present work is intended to extend the KMC part from calculations taking into account only next neighbour interactions to a more realistic description, in which also diffusion along steps and vacancy diffusion is properly described.
Sputtering: The Molecular Dynamics (MD) method is used to study sputtering of single crystal surfaces under keV ion bombardment. The main topics of the calculations are the emission of clusters and the desorption of adsorbed molecules. In addition also simulations of cluster bombardment of a surface have been performed. Experimentally in Secondary Ion Mass Spectrometry (SIMS) the emission of large (organic) molecules adsorbed on surfaces is observed for keV ion bombardment. The aim of the simulations is the determination of the processes, which cause the emission of a large intact molecule under the bombardment with a single energetic ion.
